Research
Research in the Otten group centers on molecular chemistry applied to two central themes: energy storage and catalysis.
Bipolar organic materials for redox-flow batteries
The research in the 'energy-storage' theme focuses on the development of novel charge-storage materials for redox-flow batteries (RFBs). Redox-active organic and organometallic compounds are promising alternatives to current RFB technologies based on scarce metals such as vanadium, because they consist of cheap readily available elements and can be modified to optimize their performance (redox potentials, solubilty, stability etc.). We develop RFBs with symmetric electrolyte composition, by making use of bipolar compounds as active material. This mitigates the crossover issues that currently plague many flow battery chemistries, especially in non-aqueous solvents.
We synthesize stable organic radicals and investigate how substituents alter chemical properties such as redox-potentials, solubility and stability. Based on screening by cyclic voltammetry, the most promising candidates are identified and tested in static H-cell charge/discharge experiments and in an electrochemical flow cell. We aim to combine our electrochemical characterization data with in-situ chemical analysis methods based on spectroscopy (NMR, EPR, UV/Vis) to provide detailed insight in connections between molecular structure and battery performance. This will ultimately provide flow battery chemistries that combine high cell voltage and excellent cycling longevity.
A second research line focuses on coordination and organometallic chemistry, specifically the involvement of ligand-based reactions in catalysis.
The research in the 'synthesis/reactivity' theme is broadly defined by the application of ligand design to control chemical reactions. We study coordination complexes with 'non-innocent' ligands. These ligands actively participate in (catalytic) reactions of their complexes, either by making/breaking bonds, or by functioning as electron reservior. The design of such ligands allows us to carry out unusual (redox) transformations with catalysts based on earth-abundant elements. Moreover, it provides opportunities to switch the reactivity profile (e.g., on/off, or selectivity for different substrates) through modification of oxidation- or spin-state.
Redox-active formazanate ligands
We are exploring the use of formazanate ligands in coordination chemistry and catalysis. These ligands are nitrogen-rich analogues of the well-known ß-diketiminates. Despite the fact that the parent formazans have been known for a long time, their use as ligands for the synthesis of metal complexes has not been explored in any detail. Our group has begun to uncover the similarities and differences between ß-diketiminate and formazanate ligands. We have recently shown that bis(formazanate) zinc complexes have very similar structural features as the ß-diketiminate congeners, but provide access to unusual redox-chemistry by using the ligands as electron-reservoir. The formazanate ligands in these L2Zn complexes can be sequentially and reversibly reduced to give the corresponding radical anions L2Zn- and diradical dianions L2Zn2-, which have a singlet diradical ground state. We are currently working on the synthesis of other (transition) metal complexes, with the aim to make use of the reversible redox-chemistry of the ligand framework in catalysis.
Also main group compounds with formazanate ligands participate in ligand-based redox reactions. For example, mono(formazanate)boron difluoride complexes are studied for their luminescent properties, which may be switched on/off as a function of ligand oxidation state.

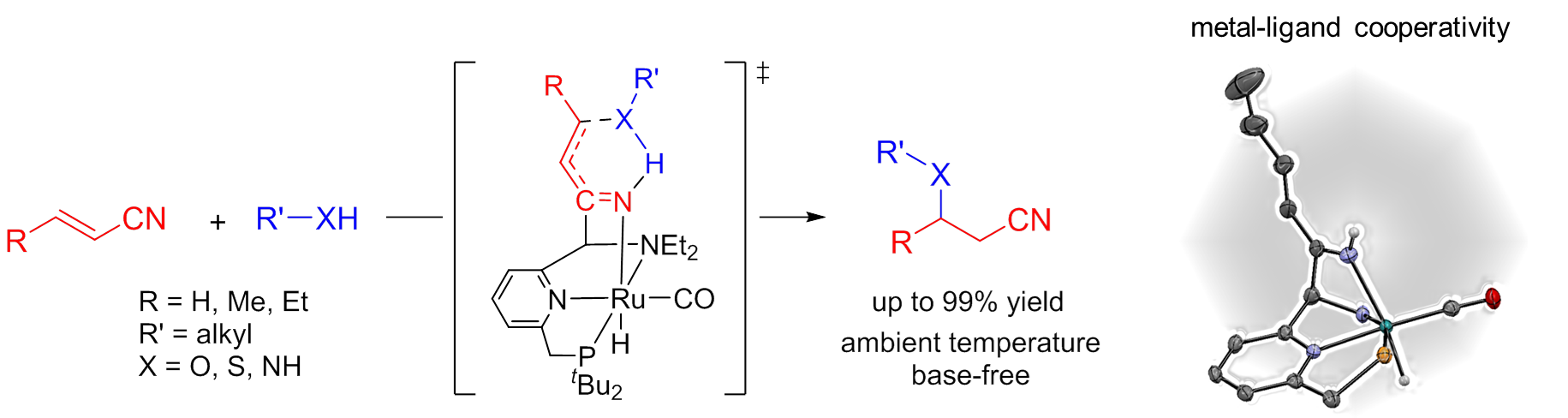
Metal-ligand cooperativity in nitrile activation: an efficient pathway for catalytic oxa-Michael addition to unsaturated nitriles
We study the reactivity of tridentate PNN pincer complexes based on Ru. These compounds, developed by Milstein and co-workers, show a wide variety of catalytic reactions that are based on aromatization/dearomatization of the pyridine fragment of the pincer backbone. We have shown that addition of nitriles to the dearomatized complex leads to formation of an addition product, in which the nitrile CN moiety forms bonds with the metal center (Ru-N) as well as the ligand (C-C). This unusual bond activation occurs with re-aromatization of the pyridine, which provides a driving force for the reaction. The isolated Ru-dieneamido product that is formed upon reaction with alkene-nitriles is shown to tautomerize to a Ru-ketimido intermediate. This Ru-N fragment (derived from the nitrile) is sufficiently basic to activate alcohols towards nucleophic attack at the C=C bond of the α‚β-unsaturated motif. The resulting oxa-Michael addition product is obtained in excellent yield/selectivity under mild reaction conditions, due to this unusal nitrile activation mechanism.
| Laatst gewijzigd: | 01 februari 2024 11:55 |