Molecular Dynamics Group

-

-
-
-
-
-
-

-
-
-
-
-
-
-

-

-
The MD group, headed by Prof. S.J. Marrink, concentrates on dynamical simulation of cellular processes. The aim is to understand and predict macroscopic behaviour of complex biomolecular systems on the basis of the effective interactions between atoms. There are several levels of approach, all of which are pursued in our group:
- For processes where quantum-dynamical aspects are important, such as proton and electron transfer, mixed quantum and classical dynamics is used;
- On the atomic level, classical molecular dynamics simulations are carried out on systems including up to hundreds of thousands of particles;
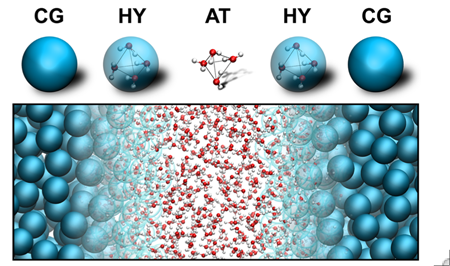
- On the molecular, but supra-atomic level, details of uninteresting atomic motions are averaged out and replaced by coarse-grained dynamics;
- For modeling of processes that require accurate treatment of a subset of degrees of freedom only, multi-scale simulation techniques are used
The MD group is home to the MARTINI coarse grained forcefield for biomolecular simulations. Furthermore, the origins of the GROMACS and GROMOS software suites are found in this group.
Last modified:21 November 2017 09.23 a.m.