Development of Multiscale Models
To take full benefit of the power of computational modeling, the resolution of the system should be tuneable. In particular, techniques that can couple the sampling speed gained with coarse-grain (CG) models to the accuracy inherent of all-atom (AA) models are needed. A straightforward way to do so is so-called backmapping, a sequential approach in which a lower-level resolution configuration is converted to a higher resolution one. More demanding is the use of hybrid methods in which multiple levels are coupled concurrently. Two types of hybid methods that are currently pioneered in the field show a lot of promise. In concurrent multiscale models, a static division of the AA and CG degrees of freedom is used, akin to the well-established QM/MM method. Alternatively, adaptive multiscale models are being developed that allow particles to change resolution during the simulation. The practical use of the above methods, however, is still limited; parameters have been optimised for specific systems only and the implementation of the methods does not yet allow large-scale applications. The challenge the Berendsen Center is facing is to develop efficient methods to seemlessly connect the different levels of resolution.
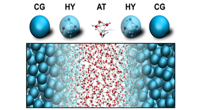
The Molecular Dynamics group actively contributes to the development of efficient multiscale methods. Recently we published a novel algorithm ('Backward') to perform backmapping transformations, based on a geometric approach that can be applied to thread atomistic models into generic coarse-grained configurations. The tool is available on the Martini website (cgmartini.nl).
In addition we pioneer the use of hybrid multiscale methods, either using virtual sites to couple the different level of resolutions, or by resorting to an adaptive resolution strategy known as the AdResS method. The latter work is done in close collaboration with Dr. Praprotnik (Univ. of Ljubljana).
Key publications:
- J. Zavadlav, M.N. Melo, S.J. Marrink, M. Praprotnik. Adaptive resolution simulation of polarizable supramolecular coarse-grained water models. J. Chem. Phys, 142:244118, 2015. abstract
- N. Goga, M.N. Melo, A.J. Rzepiela, A.H. De Vries, A. Hadar, S.J. Marrink, H.J.C. Berendsen. Benchmark of schemes for multiscale molecular dynamics simulations. JCTC, 11:1389–1398, 2015. open access
- J. Zavadlav, M.N. Melo, A.V. Cunha, A.H. De Vries, S.J. Marrink, M. Praprotnik. Adaptive resolution simulation of MARTINI solvents. JCTC, 10:2591–2598, 2014. open access
- T.A. Wassenaar, K. Pluhackova, R.A. Böckmann, S.J. Marrink, D.P. Tieleman. Going backward: A flexible geometric approach to reverse transformation from coarse grained to atomistic models. JCTC, 10:676-690, 2014. abstract
- J. Zavadlav, M.N. Melo, S.J. Marrink, M. Praprotnik. Adaptive resolution simulation of an atomistic protein in MARTINI water. J. Chem. Phys., 140:054114, 2014. open access
- T.A. Wassenaar, H.I. Ingólfsson, M. Prieß, S.J. Marrink, L.V. Schäfer. Mixing Martini: electrostatic coupling in hybrid atomistic – coarse-grained biomolecular simulations. J. Phys. Chem. B, 117:3516–3530, 2013. open access
- A.J. Rzepiela, M. Louhivuori, C. Peter, S.J. Marrink. Hybrid simulations: combining atomistic and coarse-grained force fields using virtual sites. Phys. Chem. Chem. Phys., 13:10437-10448, 2011
- A.J. Rzepiela, L.V. Sch ä fer, N. Goga, H.J. Risselada, A.H. de Vries, S.J. Marrink. Reconstruction of atomistic details from coarse grained structures. J. Comp. Chem., 31:1333-1343, 2010. abstract