EC:3.2.1.52
ß-Hexosaminidase-A (aandoening: GM2-2 Gangliosidosis, Tay Sachs)
Gangliosiden zijn glycosphingolipiden, bestaand uit een hydrofoob ceramide (N-acylsphingosine), en een hydrofiele oligosaccharide-keten, die één of meer siaalzuur-residuen bevat. In adult menselijk hersenweefsel komen tenminste twaalf verschillende gangliosiden voor, waarvan er vier (GM1, GD1A, GD1B, en GT1B) meer dan 90% van het totaal beslaan.
Deze vier types hebben allen dezelfde tetrasaccharide-keten, waaraan één (GM1), twee (GD1A en GD1B), of drie (GT1B) siaalzuur-residuen zijn gebonden.Gangliosiden zijn karakteristieke componenten van de buitenste laag van de plasmamembraan bij dierlijke cellen. Waarschijnlijk zijn ze nauw betrokken bij cel-differentiatie en cel-cel interactie. Bepaalde gangliosiden doen dienst als bindingsplaatsen voor virussen en bacteriële toxinen, of als co-receptoren voor hormonen, groeifaktoren en interferonen. De grootste gangliosiden-concentratie bevindt zich in de grijze hersenstof, waar ze 6% van de lipiden uitmaken.
Gangliosiden worden in een zuur milieu in de lysosomen van de cel, afgebroken door exohydrolases.Deze afbraak gebeurt stapsgewijs, beginnend bij het hydrofiele einde van een molekuul. Bijna elk van deze afbraakstappen kan gestoord zijn, resulterend in vetstapeling; alle bekende ganglioside-stapelingsziekten zijn het gevolg van gestoord lysosomaal katabolisme.
Defecten in ganglioside-afbraak leiden tot ernstige gevolgen voor het zenuwstelsel, doordat neuronen gevuld raken met vet-gestapelde lysosomen ; slechts kleine hoeveelheden van de gestapelde vetten verlaten de cel; zij kunnen gedetecteerd worden in cerebrospinale vloeistoffen en urine.
Al in 1936 was het bestaan van het lysosomale enzym N-acetyl-b-D-glucosaminidase (=b-hexosaminidase) bekend. In 1968 onderscheidden Robinson en Stirling bij hexosaminidase afkomstig uit humane milt, twee iso-enzymen, namelijk een zure vorm; hexosaminidase A, en een basische vorm; hexosaminidase B.
Beide enzymen zijn oligomeren, opgebouwd uit a- en b-subunits. Hexosaminidase A is opgebouwd uit zowel a- als b-subunits, hexosaminidaseB bestaat alleen uit b-subunits.
Later werd in weefsels van Sandhoff-patiënten (deze patiënten missen zowel hexosaminidase -A als -B), nog een derde iso-enzym ontdekt. Dit derde enzym, hexosaminidase S, bevat geringe katalytische activiteit, en is een dimeer bestaande uit enkel a- subunits.
In menselijk weefsel breekt hexosamininidase-A, ganglioside GM2 af, door het terminale N-acetylgalactosaminyl-residu van GM2 af te splitsen.
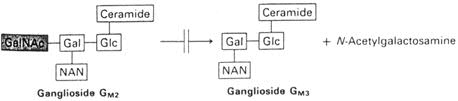
Een defect in, of deficiëntie van de a-unit van hexosaminidase-A verhindert de vorming van intact hexosaminidase A (en van het minder belangrijke hexosaminidase S), terwijl uit de niet-aangedane b-units wel intact hexosaminidase B gevormd kan worden. Biochemisch wordt hexosaminidase A-deficiëntie gekarakteriseerd door de aanwezigheid van normale, of zelfs verhoogde hexosaminidase-B concentraties. Om deze reden wordt dit defect ook wel aangeduid met de naam 'variant B van GM2-gangliosidose' of 'GM2-2 gangliosidose'. De benaming 'ziekte van Tay-Sachs' wordt gewoonlijk gebruikt voor de infantiele vorm van deze enzymatische variant. Tay-Sachs is autosomaal-recessief overerfelijk. Bij Joods-Amerikanen is 1 op de 30 personen drager, terwijl bij niet-Joodse Amerikanen slechts 1 op de 300 personen dat is; de incidentie van deze ziekte bij Joods-Amerikanen is hierdoor zo'n 100 maal groter.
Klinisch zijn er bij GM2 gangliosidose vier varianten te onderscheiden;
-
1. Infantiele Tay-Sachs.
Hierbij openbaart de ziekte zich op een leeftijd van 3 tot 5 maanden, met milde symptomen van motore zwakte. Hierna ziet men gewoonlijk een progressieve motore-mentale retardatie en verlies van gezichtsvermogen, gevolgd door macrocephalie op een leeftijd van 1-1? jaar.Uiteindelijk resulteert dit in een volledig non-responsief vegetatief stadium, en patiënten met deze vorm van GM2 gangliosidose worden gewoonlijk niet ouder dan 2 tot 4 jaar.
-
2. Juveniele GM2 gangliosidose (ziekte van Bernheimer-Seitelberger).
Symptomen van motore coördinatiestoornissen openbaren zich tussen het tweede en zesde levensjaar, gevolgd door progressieve dementie, spraakverlies en toenemende spasticiteit voor het tiende levensjaar. Over het algemeen treedt gezichtsverlies in een veel later stadium op dan bij de Tay-Sachs variant.Op 10-12 jarige leeftijd wordt gewoonlijk een vegetatief stadium bereikt, gevolgd door de dood op 10- 15 jarige leeftijd.
-
3. Chronische GM2 gangliosidose.
Men ziet tussen 2- en 5 jarige leeftijd veelal de eerste symptomen in de vorm van afwijkend lopen en een vreemd postuur. Mentaal en verbaal zijn er geen problemen, hoewel patiënten vaak emotioneel labiel zijn. Cerebrospinale problemen komen tot uitdrukking in spasticiteit, dysartrie (uitspraakstoornis t.g.v. een neurologische aandoening), coördinatiestoornissen van ledematen en romp, en toenemende spierzwakte. patiënten kunnen een leeftijd van 30-40 jaar bereiken.
-
4. Adult-onset GM2 gangliosidose.
Deze variant wordt gekenmerkt door een grote variabiliteit in klinische expressie. Het meest voorkomend zijn symptomen ten gevolge van cerebrospinaal dysfunktioneren. Onderscheid tussen de chronische en adult-onset vorm is moeilijk, omdat veel adult-onset patiënten op het moment dat ze gediagnostiseerd worden, al vele jaren (soms al sinds kinderleeftijd) vage symptomen vertoonden.
Omim
Principe van de enzymassay:
Het patiëntenmateriaal wordt geïncubeerd met het synthetische substraat 4methylumbelliferyl-β-glucosamine-N-acetyl-6-sulfate; door de werking van hexosaminidase A wordt 4methylumbelliferon (4MU) afgesplitst.De hoeveelheid 4MU die zodoende vrijkomt, is een maat voor de enzymaktiviteit, en is fluorometrisch te bepalen.
Gene review
Benodigd materiaal: leukocyten of fibroblasten.
Referenties:
-
Aanpassing op: Genetic Metabolic Diseases, Galjaard H., Elsevier/North Holland Biomedical Press,p825-826, ISBN 0-444-80143-X, 1980.
-
Gravel RA, Kaback MM, Proia RL, Sandhoff K, Suzuki K, Suzuki K. The Gm2 gangliosidoses. In:Scriver CR, Beaudet AL, Sly WS, Valle D, eds.The Metabolic and Molecular Bases of Inherited Disease. 2001; 8 edtion: 3827-3877
-
Stryer, L., Biochemistry, 4th ed., W.H. Freeman and Company, USA, 1995.