EC: 3.2.1.20
α- Glucosidase (zure maltase) (aandoening:GSDII, Morbus Pompe)
(natural substrate)
Glycogenose type II is een gegeneraliseerde glycogeenstapelingsziekte, die in 1932 ontdekt werd door Pompe.
De ziekte wordt veroorzaakt door een deficiëntie van het lysosomale enzym α- 1,4-glucosidase. Dit enzym speelt een belangrijke rol bij de afbraak van glycogeen tot glucose; het hydrolyseert de α- 1,4 en de α- 1,6 verbindingen. α- 1,4-Glucosidase komt tot expressie in verschillende weefseltypes,waaronder leukocyten en fibroblasten.
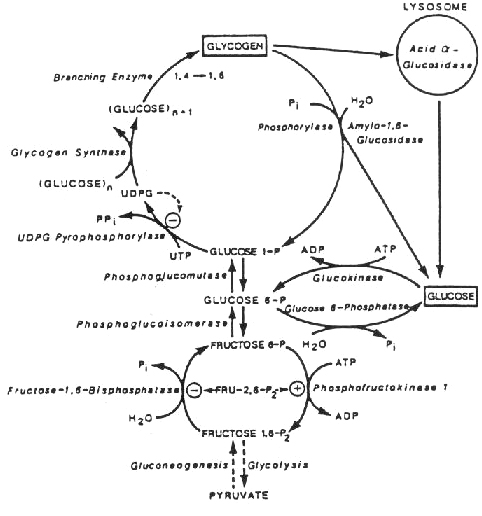
Glycogen and related metabolism in the liver Glycogenose type II wordt gekenmerkt door ernstige hypotonie en hartfalen. Bij de klassieke infantiele vorm manifesteren de symptomen zich soms al vlak na de geboorte, maar gewoonlijk ziet men na ongeveer twee maanden de eerste verschijnselen.Snelle progressie van de ziekte leidt meestal nog voor het eerste levensjaar tot de dood ten gevolge van cardio-respiratoir falen. Bij de juveniele of adulte vorm van glycogenose II, kunnen de symptomen zich op elk moment tussen kinder- en volwassen leeftijd openbaren.In eerste instantie ondervindt de patient moeilijkheden bij het bewegen en lopen. Progressieve spierzwakte zal uiteindelijk ook het funktioneren van andere skeletspieren beperken (limbgirdle disease).Bij de mildere vormen zijn het hart en andere inwendige organen niet aangetast, hoewel sommige patiënten met toenemende leeftijd toch cardiorespitatoire klachten krijgen.Vaak wordt de juiste diagnose niet onmiddellijk gesteld, omdat dit ziektebeeld gemakkelijk te verwarren is met progressieve spierdystrofie of polymyositis.
Omim
Principe van de enzymassay: Bij deze bepaling vindt incubatie van het patientenmateriaal met een vastgestelde hoeveelheid glycogeen plaats, waarna fotometrisch bepaald wordt hoeveel glucose er gevormd is tijdens de incubatie. De hoeveelheid glucose is een maat voor de activiteit van α-1,4-glucosidase.
Benodigd materiaal: leukocyten, fibroblasten of spier
Referenties: Hirschhorn R, Reuser AJJ, Glycogen Storage Disease Type II: Acid a-Glucosidase(Acid Maltase) Deficiency. In:Scriver CR, Beaudet AL, Sly WS, Valle D, eds.The Metabolic and Molecular Bases of Inherited Disease. 2001; 8 edtion: 3389-3421 Galjaard, H., Genetic Metabolic Diseases, Elsevier/North-Holland Biomedical Press, Amsterdam Nl, 1980, ISBN 0-444-80143-X. Koster, J.F. et. al., The use of leucocytes as an aid in the diagnosis of a variant of glycogen storage disease type II (Pompe's disease). Eur. J. Clin. Invest. 1972; 2 (6): 467-71.
a-Glucosidase (zure maltase) (aandoening: Morbus Pompe) (synthetisch substraat)
Principe van de enzymassay: Het patiëntenmateriaal wordt geïncubeerd met het synthetische substraat 4-methylumbelliferyl-α- D- glucopyranoside; door de werking van α- glucosidase wordt 4-methylumbelliferon (4MU) afgesplitst.De hoeveelheid 4MU die zodoende vrijkomt is een maat voor de enzymaktiviteit en is fluorometrisch te bepalen.
Benodigd materiaal: fibroblasten.