EC.2.3.1.9
3-oxo(keto)acyl-coA-thiolase
Mitochondriele en cytoplasmatische thiolase met substraat acetoacetyl-CoA
Theorie
Ketonlichamen (acetoacetaat en D3-hydroxybutyraat) worden gevormd bij overmatig vetafbraak, vasten of diabetus. Ze zijn belangrijk als oxidatieve substraten voor perifeer weefsel, als lipogene voorlopers en als regulatoren voor het metabolisme. Hun serumconcentratie wordt bepaald door het evenwicht tussen de aanmaak in de lever en het verbruik in perifeer weefsel. Een stijging in deze concentratie kan leiden tot ketoacidose en kan veroorzaakt worden door verhoogde aanmaak (bv. diabetus), verminderd verbruik en verandering in ketonlichaam-metabolisme.
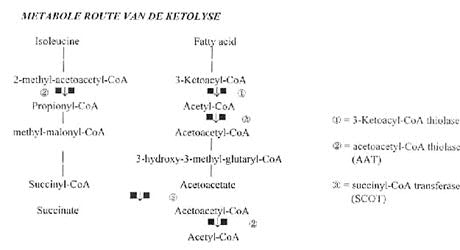
In ons lichaam wordt acetoacetaat (voorganger van acetoacetyl-CoA) als normale brandstofbron gebruikt, zo hebben de hartspieren zelfs de voorkeur voor deze stof boven glucose.
Principe van de enzymassay:
De reactie die de mitochondriële en de cytoplasmatische thiolases katalyseren gaat als volgt:
Acetoacetyl-CoA + CoA ----------> 2 acetyl CoA
Bij de activiteitsbepaling van beide thiolases wordt de afname van acetoacetyl-CoA in de aanwezigheid van CoenzymA spectrofotometrisch bepaald bij 303 nm. De cytoplasmatische thiolase is niet gevoelig voor kaliumionen, in tegenstelling tot de mitochondriële thiolase die specifiek is voor acetoacetyl CoA die door kaliumionen wordt gestimuleerd. De enzymactiviteiten verhouden zich bij gezonde controle’s als:
activiteit (mit. + cyt.) = met kaliumionen / activiteit cyt. = zonder kaliumionen ---------> 2/1
Is er sprake van een cytoplasmatische thiolase deficiëntie dan vinden we zonder kaliumionen niet of nauwelijks activiteit, omdat de mitochondriële thiolase zonder kaliumionen nauwelijks activiteit geeft. De verhouding wordt dus veel groter dan twee. Is er sprake van een mitochondriële thiolase deficiëntie dan vinden we geen stimulering door kaliumionen en zal de verhouding ongeveer 1 zijn.
Benodigd materiaal: fibroblasten
Referenties:
-
R.J.A. Wanders, L. IJlst, A.H. van Gennip, C. Jakobs, J.P. de Jager, L. Dorland, F.J. van Sprang and M. Duran, Long-chain 3-Hydroxyacyl-CoA dehydrogenase deficiency: Identification of a new inborn error of mitochondrial fatty acid b-oxidation, J.Inher.Metab.Dis. 13, 311-314 (1990).
-
R.J.A. Wanders, L. IJlst, F. Poggi, J.P. Bonnefont, A. Munnich, M. Brivet, D. Rabier and J.M. Saudubray, Human trifunctional protein deficiency: a new disorder of mitochondrial fatty acid b-oxidation, Biochemical and Biophysical research communications, vol 188, no 3, 113-1145 (1992).
-
B. Middleton, The acetoacetyl-Coenzym A Thiolases of Rat Brain and their Relative Activities during Postnatal Development, Biochem. J. 132, 731-737 (1973).
-
J.M. Saudubray, N. Specola, B. Middleton, . Lombes, J.P. Bonnefont, C. Jakobs, A. Vassault, C. harpentier, R. Day, Hyperketotic States Due to Inherited Defect of Ketolysis, Enzyme 38: 80-90 (1987).
-
J.V. Leonard, B. Middleton, J.W.T. Seakins, Acetoacetyl CoA Thiolase Deficiency Presenting as Ketotic Hypoglycemia, Pediatric Research Vol. 21 No. 2: 211-213 (1987).
-
K.E. Niezen-Koning, R.J.A. Wanders, J.P.. Ruiter, G. Visser, W.C.C. Reitsma-Bierens, H.S.A. Heymans, D.J. Reijngoud, G.P.A. Smit, Succinyl-CoA acetoacetate transferase deficiency: identification of the second patient with a neonatal onset presenting an uncomplicated clinical course, Chapter 6.2, p 79-83.
-
L. Stryer, Biochemistry, 3th ed., USA, p. 479.
-
Yamaguchi e.a., J.Clin.Invest 81, 813-817 (1988).
-
Wajner e.a., Clin Genet 41, 202-205 (1992).
-
Mitchell GA, Fukao T., Inborn errors of ketone body metabolism. In:Scriver CR, Beaudet AL, Sly WS, Valle D, eds.The Metabolic and Molecular Bases of Inherited Disease. 2001; 8 edtion:2327-2357