Research
Currently we are pursuing the following research projects in our group:
I. On-the-fly quantum dynamics in a complex environment
II. PySurf: A Framework for Database Accelerated Direct Dynamics
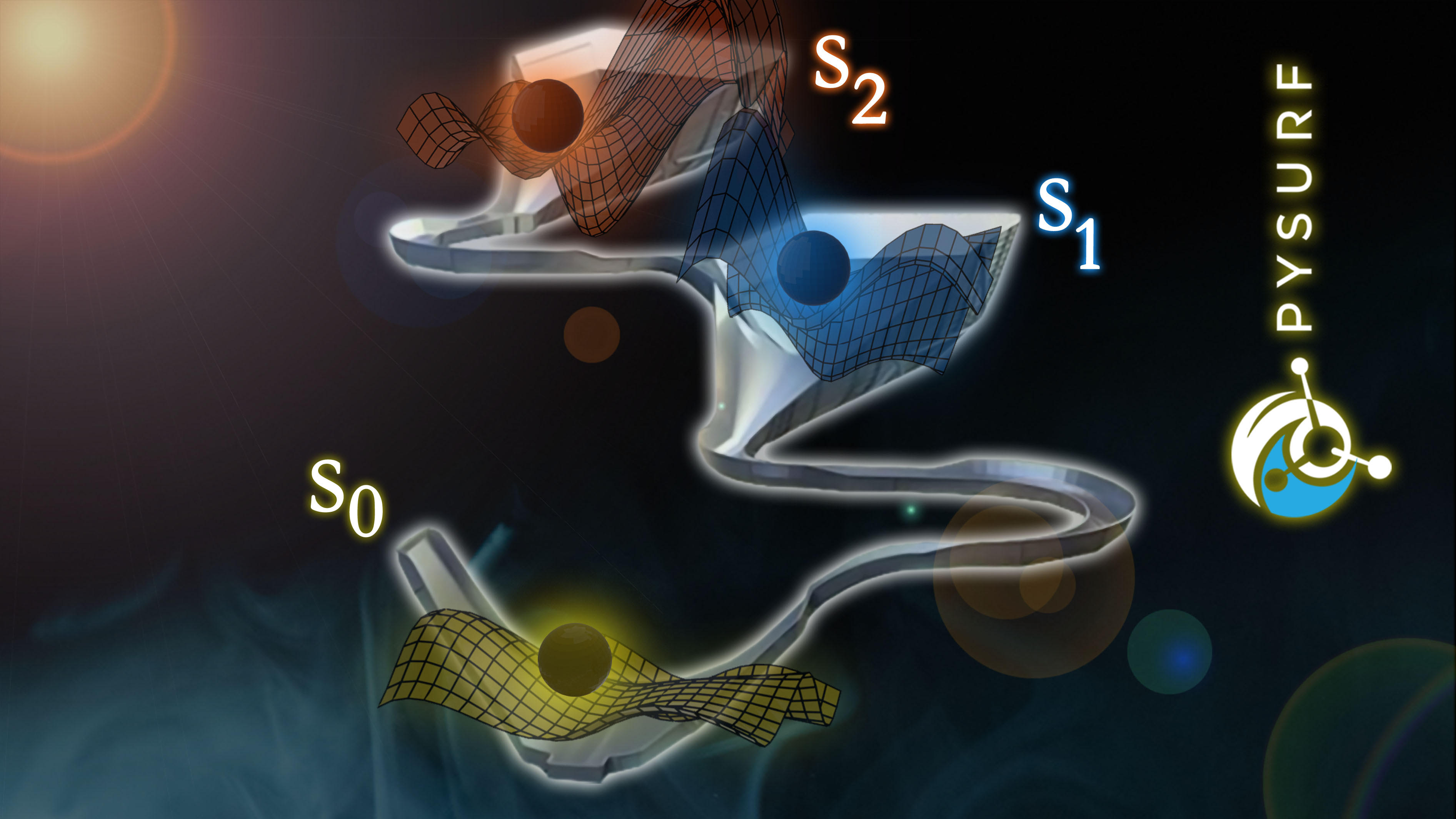
The greatest bottleneck to study photo-induced processes is computationally expensive electronic structure calculations. With PySurf, we present a novel innovative code framework to tackle the latter. It is especifically designed for rapid prototyping and development in computational chemistry. Results from ab-initio calculations are automatically preserved through a database framework and can be used e.g. for the training of machine learning models. This enables the study of (non-adiabatic) dynamics and potential energy exploration in small to medium sized systems.
III. Machine Learning in Excited-State Dynamics
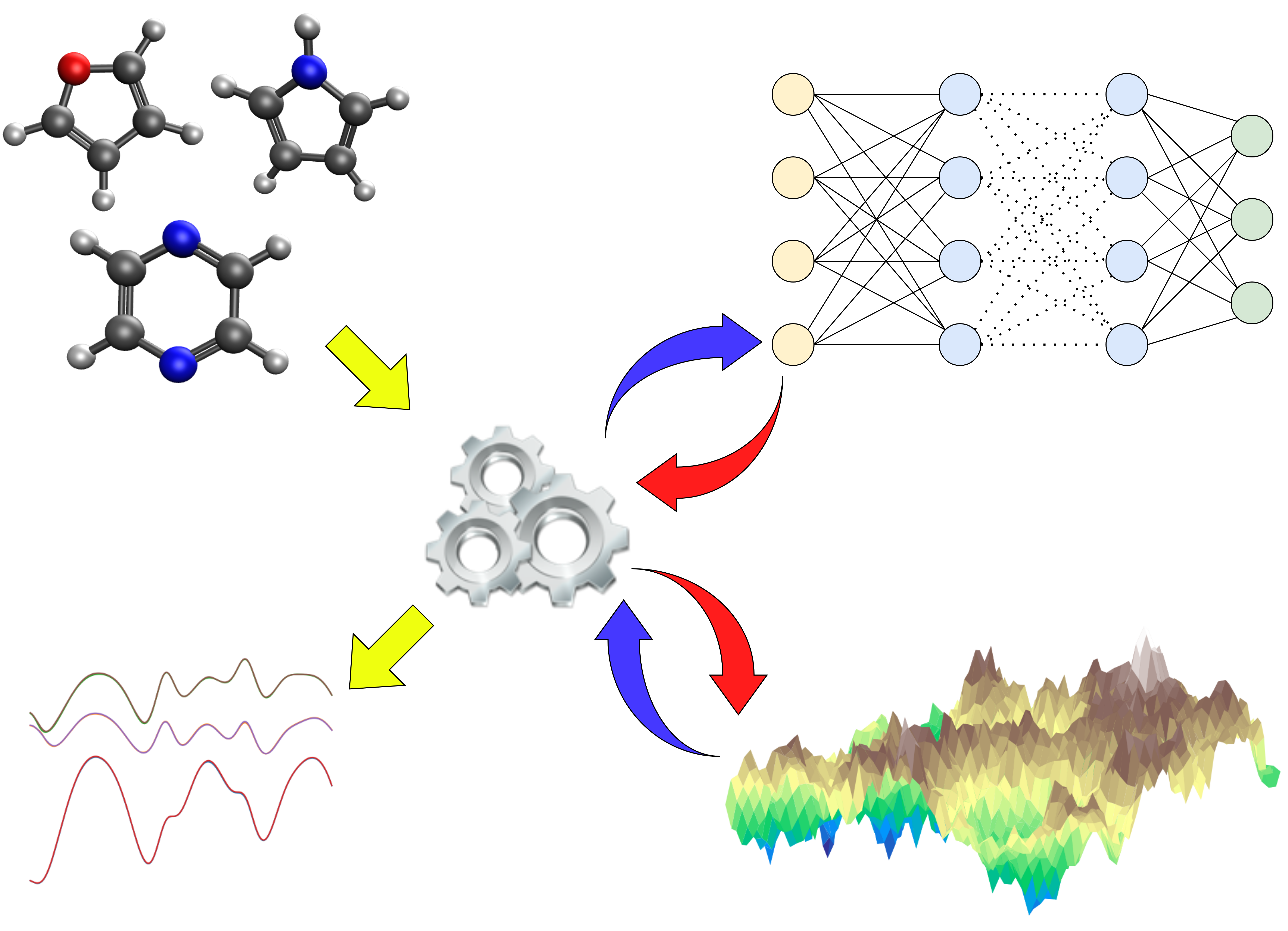
Machine learning and methods borrowed from the realm of artificial intelligence have the potential to advance the field of excited-state dynamics by significantly reducing the number of electronic structure calculations. In this project we create an extendable package to create neural network models from excited-state trajectories. We implement hyper parameter optimization through genetic algorithms, simulated annealing, grid search, random search and finite difference gradient descent. By performing comparative analysis of both hyper optimization and training size, we aim to learn more about the relationship between neural network model parameters and the molecules we try to model in this way.
IV. Photo-switchable DNA G-quadruplex; using hybrid quantum mechanics/molecular mechanics approach
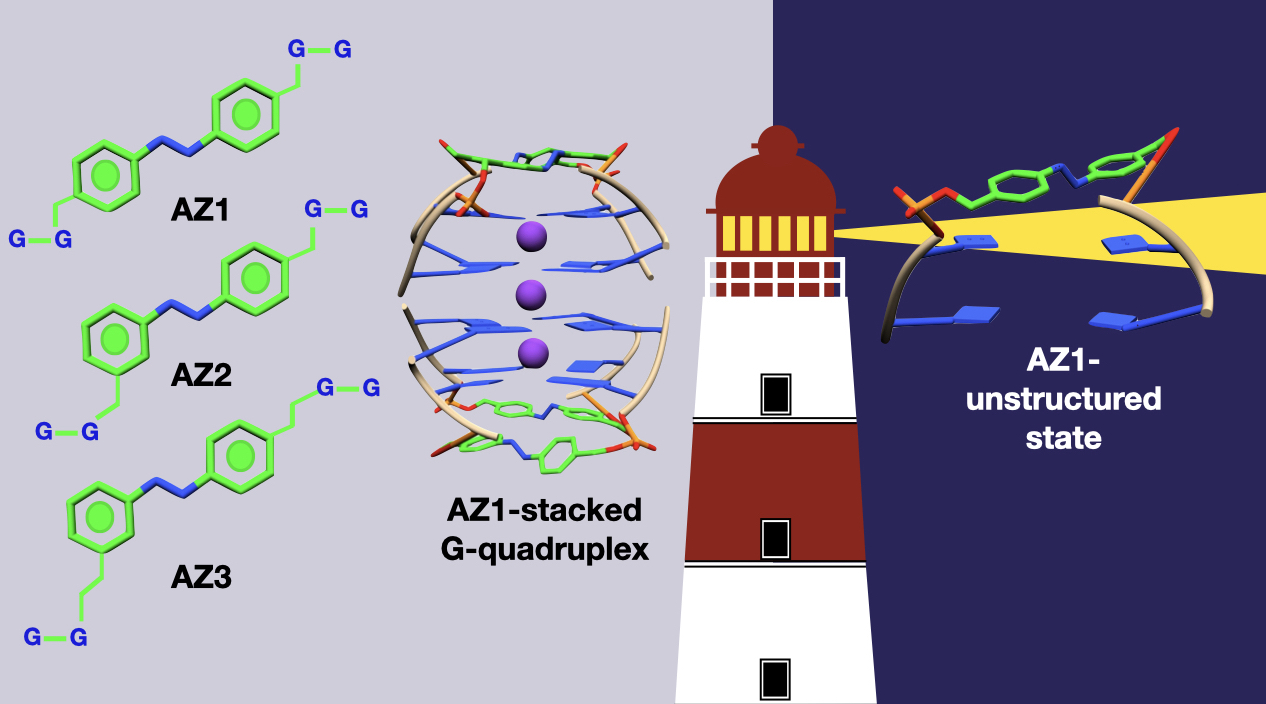
Introducing photoswitches into the DNA G-quadruplex provides excellent opportunities to control folding and unfolding of these assemblies, demonstrating their potential in the development of novel nanodevices with medical and nanotechnology applications. Using molecular dynamics in combination with hybrid quantum mechanics and molecular mechanics, we provide atomistic insights into the photoresponsive formation of photoswitchable G-quadruplex motifs, thus providing design principles for developing azobenzene-based photocontrollable DNA G-quarduplexes.
V. Photo-switchable molecular motors; using surface hopping dynamics
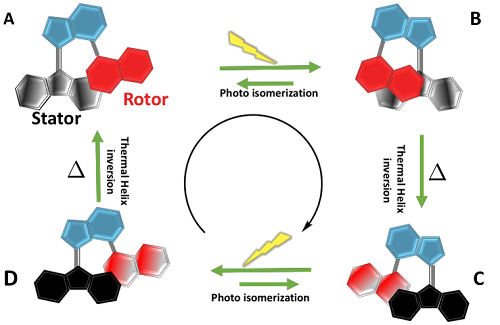
Light-driven molecular- motors and switches that can exist in two or more distinct (meta)stable states, are excellent candidates for developing modern technologies utilizing light, such as nanomachines or molecular electronic devices. Using advanced electronic structure methods, we explore the excited-state dynamics and model relevant spectra to translate experimental observation into an atomic-level mechanism picture providing detailed insight into the primary photo-chemical reactions.
VI. Excited-state processes in far-red near IR fluorescent proteins; using hybrid quantum mechanics/molecular mechanics approach
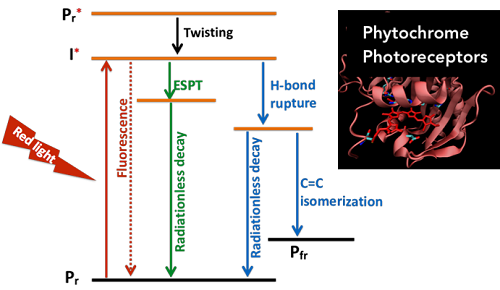
Far-red and near IR fluorescent proteins offer unique opportunities for revolutionizing next-generation light induced technologies such as optogenetics and photopharmacology. The protein environment plays a crucial role in structural stabilization and the excited-state processes of the chromophore. We use hybrid quantum mechanics/molecular mechanics to investigate the influence of the environment in excited-state processes of the chromophore to engineer the protein/chromophore structure for a tailored excited-state process matching specific application.
VII. Singlet fission in molecular solids; using non-orthogonal configuration interaction approach
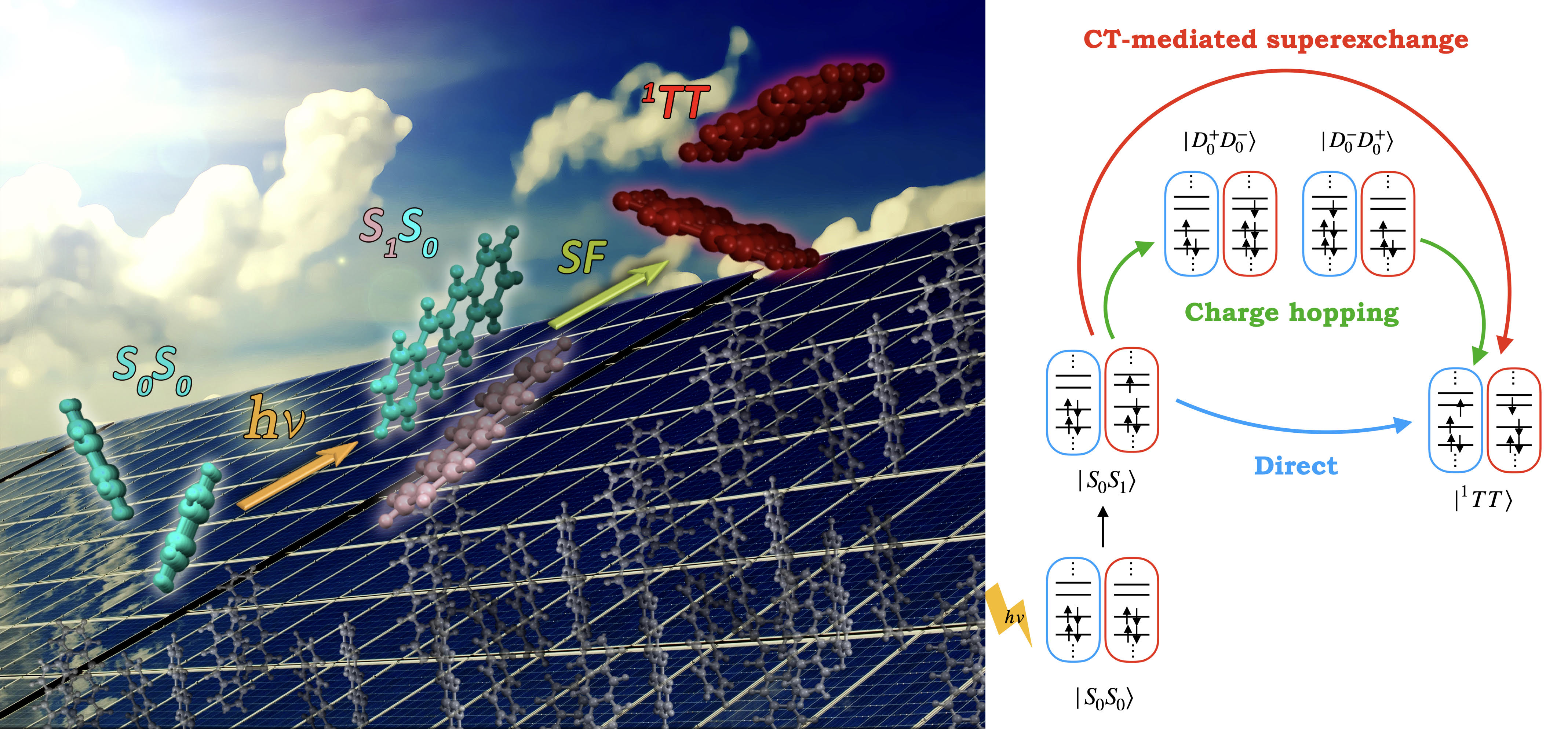
Singlet fission (SF) has been proposed as an alternative to boost solar cells’ efficiency since, in principle, two pairs of charge carries can be generated per single absorbed photon. In this process, two singlet-coupled triplets (1TT) are formed from a photo excited chromophore combined with a neighbor in the ground state (S1S0). The theoretical modelling of SF is often done by employing a gas-phase dimer model which has shown its utility as a first approximation to explain the process. However, it does not always cover all the physics and the effect of surrounding molecules has to be included in such cases. In this project, we explore how the crystal packing (environment) influences the SF process by means of analyzing the electronic couplings and the excited character of the molecules inside the crystal structure.
| Last modified: | 09 November 2021 11.52 p.m. |